The CEM/AKB4 cells were hypersensitive to the Aurora A inhibitor MLN8237. CEM/AKB4 cells were, however, cross resistant to a selective Aurora B inhibitor, AZD1152, indicating an Aurora B dependant mechanism of resistance. Although ZM447439 is known to inhibit Aurora A we excluded the possibility of an Aurora A dependent mechanism contributing to resistance to these cells by the lack of Aurora A gene and protein alterations in CEM/AKB4 cells and a lack of cross resistance to the selective Aurora A inhibitor MLN8237. This is in agreement with other reports that show the cytotoxic activity of ZM447439 is mediated through Aurora B, not Aurora A inhibition. Detection of a G160E point Trichostatin A mutation in the kinase domain of Aurora B suggested that resistance in CEM/ AKB4 cells is mediated through impaired binding of the drug to the target kinase. Genetic alterations to drug targets are common mechanisms mediating resistance to targeted therapies; point mutations in BCR-ABL conferring resistance to Imatinib in leukaemia is a classic example. Moreover, the G160E mutation in Aurora B has been reported in colorectal cells selected for resistance to ZM447439. Our findings in a leukaemia cell line further validate that the 160 position is particularly important for drug binding and that point mutations of this residue afford highly penetrant resistance. This mutation should be validated in a clinical setting as it may be important in the use of Aurora B inhibitors and resistance to therapy, much as the T315I BCR-ABL mutation is highly prognostic of outcome for Imatinib treatment in CML patients. As yet, the G160E mutation has not been reported in studies of Aurora B inhibitors in animal models or clinical studies. Although the Aurora B G160E substitution has been shown to independently confer resistance to Aurora B inhibitors it has not been conclusively shown how drug binding is affected. We therefore employed a molecular modelling approach to understand how the G160E substitution alters drug binding and to gain further insights into drug-target interactions of Aurora B inhibitors. Our docking results confirm that binding of ATP to Aurora B is unaltered in mutant Aurora B compared to the wildtype, thereby maintaining catalytic 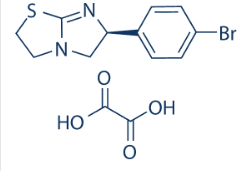 activity. We showed that hydrogen bonding of Aurora B inhibitors to the Ala173 and Lys122 residues are key interactions mediating drug activity by preventing catalytic binding of ATP. However, the presence of the G160E mutant hinders the ability of inhibitors to penetrate as far into the binding pocket as the wild-type enzyme precluding the formation of these hydrogen bonds. Presumably inhibitors are only able to bind to the mutant enzyme in modes that do not compete effectively with ATP and substrate binding, thereby allowing catalytic activity in the presence of the drug and a resistant phenotype. It would be Adriamycin expected that any Aurora B inhibitor that has a similar active binding motif would be affected, explaining the cross resistance of cells with this mutation to structurally related inhibitors in our studies and others. Our models could therefore be used as a screen to identify, or rationally design, inhibitors with novel binding modes that may abrogate Aurora B G160E mediated resistance. The progression of resistance with repeated or higher concentration drug exposure is an important consideration in the treatment of relapsed disease. Both CEM/AKB8 and CEM/AKB16 cells showed a dose dependent increase in transcriptional activity of MDR1, however P-glycoprotein was not functionally active in either case.
activity. We showed that hydrogen bonding of Aurora B inhibitors to the Ala173 and Lys122 residues are key interactions mediating drug activity by preventing catalytic binding of ATP. However, the presence of the G160E mutant hinders the ability of inhibitors to penetrate as far into the binding pocket as the wild-type enzyme precluding the formation of these hydrogen bonds. Presumably inhibitors are only able to bind to the mutant enzyme in modes that do not compete effectively with ATP and substrate binding, thereby allowing catalytic activity in the presence of the drug and a resistant phenotype. It would be Adriamycin expected that any Aurora B inhibitor that has a similar active binding motif would be affected, explaining the cross resistance of cells with this mutation to structurally related inhibitors in our studies and others. Our models could therefore be used as a screen to identify, or rationally design, inhibitors with novel binding modes that may abrogate Aurora B G160E mediated resistance. The progression of resistance with repeated or higher concentration drug exposure is an important consideration in the treatment of relapsed disease. Both CEM/AKB8 and CEM/AKB16 cells showed a dose dependent increase in transcriptional activity of MDR1, however P-glycoprotein was not functionally active in either case.