Although the two mutations in the basic amino acids in p23 did not affect virus replication, they could disrupt the nuclear localization of p23 and prevent the entry of HCRSV genome into the nucleus. It is believed that vir-miRNAs play essential roles in overcoming host resistance for efficient infection of viruses. When the p23 is unable to enter nucleus, it will not be able to interact with host genomes and thus interferes with host transcription that confers resistance. In addition, the lack of vir-miRNAs generated by the p23 sequence would not be made available to target host sequences to counteract host defence. In this study, we have adopted two approaches to uncover additional function of p23. First, a AbMole Gambogic-acid transgenic approach was used to study the function of amiRp23 to silence its target p23 gene. The amiRp23 was designed specifically to cleave p23 messenger RNA or to inhibit p23 translation. It may also downregulate viral gRNA in the p23 region, but not other regions of the viral genome. The sgRNAs and viral genes located outside the p23 gene region are not affected. Therefore, the downregulation of p23 using 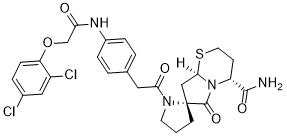 amiRp23 caused silencing of p23 and inhibition of long distance virus movement and resulted in reduced symptom severity in infected leaves. Since the kenaf plants grown under our laboratory conditions did not produce flowers, we were unable to generate transgenic progeny plants. However, using the apical meristem inoculation method, we have introduced AbMole Alprostadil amiRNA into the plants to silence p23. Due to these limitations, we were also unable to generate a reporter transgene to monitor the silencing efficiency of amiRp23. Another approach is the use of the single molecule detection technique FISH to study virus replication. It is a direct visualization technique which tracks signals in a single cell. It is a more specific and sensitive method for study of viral replication, as compared to Northern blot which requires relatively larger amount of RNA samples. We chose this method over northern blot due to the lack of sufficient amount of RNAs present in some of our test samples. Therefore, the FISH technique can be broadly applied to other virus replication studies. In conclusion, this study has uncovered an additional function of p23 of HCRSV. Although the basic amino acid mutants of p23 are prevented from entering the nucleus, those mutants are still able to replicate but unable to be moved long distance. This indicates yet another example of multi-functional roles of plant viral genes. The known functions of miRNAs include a range of biological processes, including cell differentiation, proliferation, apoptosis, and tissue development. In particular, miRNAs play critical roles in regulating osteogenic differentiation of mesenchymal stem cells. For example, miR-138, miR-204, and miR-20a have been reported to regulate osteoblast differentiation by targeting various osteoblast genes. However, more evidence for the roles of miRNAs in regulating osteogenic differentiation is needed. In view of the importance of miRNAs in the regulation of osteoblast differentiation, we wondered whether the Wnt/bcatenin pathway might be regulated by miRNAs during osteoblast differentiation of hBMSCs. We performed microarray analysis and identified miR-346 as a noncoding RNA that directly binds to the 39-untranslated region of GSK-3b mRNA. The aim of the present study was to validate the regulatory relationship between miR-346 and GSK-3b in hBMSCs and to investigate the role of this mechanism in osteogenic differentiation.
amiRp23 caused silencing of p23 and inhibition of long distance virus movement and resulted in reduced symptom severity in infected leaves. Since the kenaf plants grown under our laboratory conditions did not produce flowers, we were unable to generate transgenic progeny plants. However, using the apical meristem inoculation method, we have introduced AbMole Alprostadil amiRNA into the plants to silence p23. Due to these limitations, we were also unable to generate a reporter transgene to monitor the silencing efficiency of amiRp23. Another approach is the use of the single molecule detection technique FISH to study virus replication. It is a direct visualization technique which tracks signals in a single cell. It is a more specific and sensitive method for study of viral replication, as compared to Northern blot which requires relatively larger amount of RNA samples. We chose this method over northern blot due to the lack of sufficient amount of RNAs present in some of our test samples. Therefore, the FISH technique can be broadly applied to other virus replication studies. In conclusion, this study has uncovered an additional function of p23 of HCRSV. Although the basic amino acid mutants of p23 are prevented from entering the nucleus, those mutants are still able to replicate but unable to be moved long distance. This indicates yet another example of multi-functional roles of plant viral genes. The known functions of miRNAs include a range of biological processes, including cell differentiation, proliferation, apoptosis, and tissue development. In particular, miRNAs play critical roles in regulating osteogenic differentiation of mesenchymal stem cells. For example, miR-138, miR-204, and miR-20a have been reported to regulate osteoblast differentiation by targeting various osteoblast genes. However, more evidence for the roles of miRNAs in regulating osteogenic differentiation is needed. In view of the importance of miRNAs in the regulation of osteoblast differentiation, we wondered whether the Wnt/bcatenin pathway might be regulated by miRNAs during osteoblast differentiation of hBMSCs. We performed microarray analysis and identified miR-346 as a noncoding RNA that directly binds to the 39-untranslated region of GSK-3b mRNA. The aim of the present study was to validate the regulatory relationship between miR-346 and GSK-3b in hBMSCs and to investigate the role of this mechanism in osteogenic differentiation.
The shotgun proteome technology becomes powerful in large-scale proteomic analyses
Various proteome profiles of silkworm tissues have been established covering head, integument, midgut, fat baby, embryos, peritrophic membrane, as well as some endocrine Organs. In this study, Shotgun and 2-DE followed by MALDI-TOF MS were attempted to analyze the proteome profile of silkworm MTs from Day-5 fifth instar larvae. The raw data sets were searched against the in-house silkworm database with SEQUEST algorithms. To minimize the false positive sequence matches, the FDR of the identifications were searched through a target-decoy database and further validated by TPP and APEX analysis. A total of 1,367 proteins were identified in the MTs of silkworm; they are involved in ion and water transport, metabolism, detoxification and defense mechanism. This data may contribute to a better understanding of the function of silkworm MTs. As we known, the methodology could be used to identify proteins using mass spectral data with EST dataset. However, the ESTs are short, AbMole Acetylcorynoline one-shot sequences with no overlapping sequencing and contain errors, and of course not as reliable as full genome sequences. In the present work, the proteome technology was attempted to characterize the silkworm larval MTs proteins profile. The raw data were searched against the local database which was constructed based on the predicted proteins from silkworm genome database with mass spectrometry software.Then, the FDR of the identifications were searched through a targetdecoy database and further validated by TPP and APEX analysis. The approach presented in this study can minimize the false positive sequence matches, and provide clues 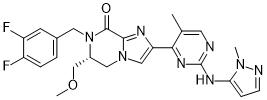 for elucidating the functions of genes underlying specific processes and identifying candidate genes predicted to regulate traits of interest. The mulberry silkworm, Bombyx mori, is a domesticated insect for silk production and a model lepidopteral insect for pest control. MTs of silkworm have important roles in excreting waste products and expelling toxins. Kajiwara et al. identified 127 proteins by MALDI mass spectrometry in silkworm MTs of fifth-instar day-3 larva. Here in our work, a total of 1,367 proteins of silkworm MTs were identified. Our proteomics study will not only enrich proteomic data of silkworm MTs, but will also offer us an important insight into understand the role of MTs in silkworm. KEGG analysis showed that these MT proteins were involved in metabolism, AbMole Nitisinone genetic information processing, environmental information processing, cellular processes and organismal systems. Especially, cytochrome P450s and glutathione transferases are highly enriched in the MTs of silkworm. Cytochrome P450s comprise a large family of genes responsible for the oxidative metabolism which involved in the metabolism of pesticides principally in the insects. In Drosophila, cytochrome P450s exhibit tissue-specific expression pattern, manipulation of Cyp6g1 in MTs could result in resistance to DDT and imperil the survival of the fly. Glutathione transferase detoxifies both endogenous and xenobiotic compounds by conjugation reactions with reduced glutathione to produce endogenous and xenobiotic compounds more easily excreted by excretory organ, such as insect MTs. McGettigan et al. found that MTs of Drosophila could sense bacterial challenge, and mount an effective killing response. In this work, we identified two antimicrobial peptides from silkworm MTs, lysozyme and lectin, which are related to immune response. The nitric oxide synthase produces nitric oxide, an immune modulator in insects. Although we could not identify the NOS from the tubule protein samples in this study, we found the NOS transcripts in the microarray data of silkworm MTs.
for elucidating the functions of genes underlying specific processes and identifying candidate genes predicted to regulate traits of interest. The mulberry silkworm, Bombyx mori, is a domesticated insect for silk production and a model lepidopteral insect for pest control. MTs of silkworm have important roles in excreting waste products and expelling toxins. Kajiwara et al. identified 127 proteins by MALDI mass spectrometry in silkworm MTs of fifth-instar day-3 larva. Here in our work, a total of 1,367 proteins of silkworm MTs were identified. Our proteomics study will not only enrich proteomic data of silkworm MTs, but will also offer us an important insight into understand the role of MTs in silkworm. KEGG analysis showed that these MT proteins were involved in metabolism, AbMole Nitisinone genetic information processing, environmental information processing, cellular processes and organismal systems. Especially, cytochrome P450s and glutathione transferases are highly enriched in the MTs of silkworm. Cytochrome P450s comprise a large family of genes responsible for the oxidative metabolism which involved in the metabolism of pesticides principally in the insects. In Drosophila, cytochrome P450s exhibit tissue-specific expression pattern, manipulation of Cyp6g1 in MTs could result in resistance to DDT and imperil the survival of the fly. Glutathione transferase detoxifies both endogenous and xenobiotic compounds by conjugation reactions with reduced glutathione to produce endogenous and xenobiotic compounds more easily excreted by excretory organ, such as insect MTs. McGettigan et al. found that MTs of Drosophila could sense bacterial challenge, and mount an effective killing response. In this work, we identified two antimicrobial peptides from silkworm MTs, lysozyme and lectin, which are related to immune response. The nitric oxide synthase produces nitric oxide, an immune modulator in insects. Although we could not identify the NOS from the tubule protein samples in this study, we found the NOS transcripts in the microarray data of silkworm MTs.
At the same time the performance of sampling method kept satisfyingly, but the performance of entrop
Furthermore, the endogenously-generated Th17 response is qualitatively distinct from that generated as a result of adoptive transfer of in vitro polarized Th17 cells, both in its sensitivity to GC and its requirement for IL-1R signaling. These data demonstrate that the Th17 response generated endogenously is substantially different than the response acquired upon adaptive transfer of in vitro polarized Th17 cells and OVA challenge, suggesting that in some instances, the outcomes of an adoptive transfer study may not be generalizable to other models of allergic airway disease. Clarification of where these models fit in terms of clinical asthma demands robust comparative analyses. As exemplified by our findings, the functional significance of the Th17 response in promoting allergic airway disease is determined by the conditions in which it was generated, ranging from protective to pathogenic. Given the heterogeneity among patients with clinical asthma, the AbMole Nitisinone contribution of a single type of immune response may also vary among patients, which should be considered during pharmacologic development and clinical testing. One of the main fields in biological researches is to reveal biological regulatory networks that control different functions. In the past, molecular interactions have been established at a rather slow pace. For example, it took more than a decade from the discovery of the well-known tumor suppressor gene p53 to the establishment of its regulatory feedback loop with the protein MDM2. This situation has been qualitatively changed thanks to the 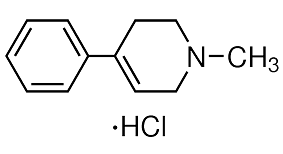 development of different new biotechnologies, especially microarray expression experiments. Accordingly, theoretical systems biology has provided several algorithms for the deduction of regulatory interactions from experimental data. These algorithms can be employed to effectively reconstruct biological regulatory networks. One of the successful network reconstruction AbMole Nitroprusside disodium dihydrate methods is reverse engineering approach, which has been advancing very rapidly in recent years. At present, there are mainly four types of the reverse engineering algorithms: correlation-based methods, information-theoretic methods, Bayesian network predictions, and methods based on dynamic models. In this paper, we apply the simplest method, the correlation-based Boolean reverse engineering, to discuss algorithms for the optimization of experiments in network construction. In this paper, we discuss a reverse engineering approach to reconstruct biological regulatory network from experiments. Instead of using real data from experiments, we applied three well-known regulatory networks as tests to generate the trajectories of the networks. We focus on how to design the experiment to get the most useful information from data of evolution trajectory. For this purpose, we propose three methods, namely, maximum distance method, trajectory entropy method, and sampling method, to derive the optimal initial conditions for experiments. The performance of these methods is discussed and evaluated by comparing the reconstructed networks with the original ones in three known systems. We also test our methods under more realistic circumstance when the number of nodes that can be perturbed is limited. From these analyses we conclude that the sampling method is the best among the three methods. Two issues are called for further investigation. The calculation cost of entropy method increases very rapidly as the node number increase; it becomes very time-consuming when the node number is large. In contrast, the calculation time of max-distance and sampling methods increase very slowly with the increase of the node number of network.
development of different new biotechnologies, especially microarray expression experiments. Accordingly, theoretical systems biology has provided several algorithms for the deduction of regulatory interactions from experimental data. These algorithms can be employed to effectively reconstruct biological regulatory networks. One of the successful network reconstruction AbMole Nitroprusside disodium dihydrate methods is reverse engineering approach, which has been advancing very rapidly in recent years. At present, there are mainly four types of the reverse engineering algorithms: correlation-based methods, information-theoretic methods, Bayesian network predictions, and methods based on dynamic models. In this paper, we apply the simplest method, the correlation-based Boolean reverse engineering, to discuss algorithms for the optimization of experiments in network construction. In this paper, we discuss a reverse engineering approach to reconstruct biological regulatory network from experiments. Instead of using real data from experiments, we applied three well-known regulatory networks as tests to generate the trajectories of the networks. We focus on how to design the experiment to get the most useful information from data of evolution trajectory. For this purpose, we propose three methods, namely, maximum distance method, trajectory entropy method, and sampling method, to derive the optimal initial conditions for experiments. The performance of these methods is discussed and evaluated by comparing the reconstructed networks with the original ones in three known systems. We also test our methods under more realistic circumstance when the number of nodes that can be perturbed is limited. From these analyses we conclude that the sampling method is the best among the three methods. Two issues are called for further investigation. The calculation cost of entropy method increases very rapidly as the node number increase; it becomes very time-consuming when the node number is large. In contrast, the calculation time of max-distance and sampling methods increase very slowly with the increase of the node number of network.
UBB may affect proinflammatory signaling in the CNS via inhibitory mechanisms of ubiquitin-dependent signaling
In a subset of preinvasive lesions, and are further lost in cancer and perineural invasion lesions. The rare tumor-associated primary cilia are shorter in preinvasive and invasive prostate cancer, which may reflect dysfunction. High nuclear ��-catenin correlates with low primary cilia frequency in normal prostate tissue. In future work, it will be important to determine if primary cilia loss is causal to tumor formation. Ubiquitylation has been well characterized to regulate vital cellular processes mainly through proteasome-dependent degradation of polyubiquitinated substrates; however, proteolysisindependent roles of ubiquitylation have emerged as key mechanisms in various signaling cascades. Typically, polyubiquitin chains that target proteins for degradation by the proteasome are linked through K48 of ubiquitin. On the contrary, K63-linked polyubiquitin chains play multiple roles in kinase activation, DNA repair and intracellular trafficking via proteasome-independent mechanisms. A frameshift mutation of ubiquitin called ubiquitin +1 was found in the aging and Alzheimer’s disease brains. UBB +1 is generated by transcriptional dinucleotide deletion within the mRNA resulting in a 19-amino acid extension at the Cterminus of ubiquitin. This mutant ubiquitin cannot link to substrates targeted for proteasomal degradation, but is ubiquitylated to form a polyubiquitin chain. Ubiquitylated UBB +1 is refractory to deubiquitination, resulting in dominant inhibition of the ubiquitin-proteasome system. Recent evidences have revealed that UBB +1 is detected as pathological hallmarks in various neurodegenerative diseases and exacerbates the proteasomal dysfunction and deposition of toxic proteins. It was also reported that UBB +1 exerts a neurotoxic effect by suppressing proteasome-dependent proteolysis in neurons. Astrocytes, the most abundant glial cells in the central nervous system, play important roles in maintaining the homeostatic environment and immune regulation, producing a repertoire of inflammatory mediators including chemokines, cytokines and adhesion molecules. Interleukin-1b and tumor necrosis factor-a serve as major regulators of immune and inflammatory responses in the CNS, and elevated expression of these cytokines occurs in injury, infection, stroke, inflammation and degenerative disorders such as AD. These proinflammatory cytokines induce 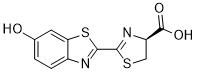 expression of multiple genes associated with inflammation by human astrocytes. In response to IL-1b and TNF-a, ubiquitylation-dependent activation of TNFassociated factor 6 and TRAF2 complexes leads to activation of TGF-b-activated kinase 1 which activates nuclear factor kappa B and c-Jun NH2-terminus kinase pathways. In this study, we investigated the effect of UBB +1 on proinflammatory signaling such as IL-1b and TNF-a in human astrocytes, and its functional relevance of ubiquitindependent kinase activation.
expression of multiple genes associated with inflammation by human astrocytes. In response to IL-1b and TNF-a, ubiquitylation-dependent activation of TNFassociated factor 6 and TRAF2 complexes leads to activation of TGF-b-activated kinase 1 which activates nuclear factor kappa B and c-Jun NH2-terminus kinase pathways. In this study, we investigated the effect of UBB +1 on proinflammatory signaling such as IL-1b and TNF-a in human astrocytes, and its functional relevance of ubiquitindependent kinase activation.
Many different mechanisms can result was hypothesized to result in increased nuclear beta-catenin
This low nuclear ��-catenin could be due to lack of canonical Wnt ligands in the tissue, or loss of pathway components needed to translocate ��-catenin into the nucleus, such as Jouberin. Interestingly, cancers surrounding nerves were shown to have increased nuclear ��catenin compared to other cancers. Perhaps the microenvironment of perineural invasion preferentially allows for pathway activation. We did see a group of prostate cancers that had high nuclear ��-catenin, and these cancers had both low percentage of ciliated cells and a decrease in cilia length. We hypothesize that shorter cilia may be dysfunctional and would fail to suppress the Wnt pathway. The low sample sizes of our cohort should be acknowledged. Our data suggests that other factors besides primary cilia may be controlling canonical Wnt signaling in the majority of prostate cancers. Upon investigation of a correlation between clinical parameters and nuclear ��-catenin, we found that high nuclear ��-catenin correlates with increased incidence of capsular penetration and that moderate/low ��-catenin correlates with increased incidence of biochemical recurrence. These results appear to be contradictory given biochemical recurrence and capsular penetration have been associated with disease-recurrence. However, several published reports on ��-catenin agree with both of our results. Chen et al. demonstrated that higher total expression of ��-catenin was found in higher tumor stage cancers versus  lower tumor stage cancers. Also in agreement with our findings, Horvath et al. investigated biochemical survival and correlated lower nuclear ��-catenin with biochemical relapse. These seemingly contradictory data, where high nuclear ��-catenin correlated with capsular penetration and moderate/low nuclear ��-catenin correlated with biochemical recurrence, may be explained by the biology of biochemical recurrence. Biochemical recurrence can occur as a result of PSA production by disseminated tumors cells that expand months following surgical removal of the primary tumor and this does not necessarily correlate with capsular penetration which is a pathologic diagnosis made at the time of surgery. This lack of correlation between biochemical recurrence and capsular penetration is also evident in our cohort. While this lack of correlation may explain our contradictory clinical correlations, further studies with a larger cohort are needed. Our findings have shown that prostate cancer cells have a very low frequency of primary cilia. An important question for therapeutics is whether this loss of primary cilia is a driver of prostate cancer. A future direction is to determine if primary cilia loss is causal in tumor formation in the prostate. If cilia are causal in cancer formation, use of drugs that result in reexpression of primary cilia in cancers may be a promising therapy. This would require the determination of the cause of primary cilia loss in prostate cancers.
lower tumor stage cancers. Also in agreement with our findings, Horvath et al. investigated biochemical survival and correlated lower nuclear ��-catenin with biochemical relapse. These seemingly contradictory data, where high nuclear ��-catenin correlated with capsular penetration and moderate/low nuclear ��-catenin correlated with biochemical recurrence, may be explained by the biology of biochemical recurrence. Biochemical recurrence can occur as a result of PSA production by disseminated tumors cells that expand months following surgical removal of the primary tumor and this does not necessarily correlate with capsular penetration which is a pathologic diagnosis made at the time of surgery. This lack of correlation between biochemical recurrence and capsular penetration is also evident in our cohort. While this lack of correlation may explain our contradictory clinical correlations, further studies with a larger cohort are needed. Our findings have shown that prostate cancer cells have a very low frequency of primary cilia. An important question for therapeutics is whether this loss of primary cilia is a driver of prostate cancer. A future direction is to determine if primary cilia loss is causal in tumor formation in the prostate. If cilia are causal in cancer formation, use of drugs that result in reexpression of primary cilia in cancers may be a promising therapy. This would require the determination of the cause of primary cilia loss in prostate cancers.