Patients with DV infection show various clinical symptoms that range from no significant illness or mild fever to life-threatening Dengue hemorrhagic fever and Dengue shock syndrome. Currently, only supportive treatments are available. Although considerable research has been AZD2281 directed towards the development of a safe and effective DV vaccine since the mid-20th century, there are no approved commercial products available. Therefore, to combat DV and other related viral diseases, it is advisable to develop novel strategies for discovering new antiviral agents. Recent progress in the biology has brought with it many protein structures for virtual screening as drug targets. However, without a previously validated target site on the targeted protein as a reference point, the number of lead candidates obtained from this type of screening is very large. Cellular toxicity further complicates biological activity assays as well. Therefore, the utilization of VS is somewhat hindered by the processes that follow, namely, the labor-intense, time-consuming verification process and the toxicity assays required for processing large amounts of lead candidates. Here, in an attempt to devise a less resource-demanding screening process, we have focused on computational approaches that are solely based on the structures of a designated region of the target protein. Then, we performed VS on a set of medical compounds because we recognized that using medical compounds could potentially minimize cellular toxicity. To reduce the number of lead candidates, we further refined the VS output by structural clustering for the identification of novel structural characteristics. Compounds with novel structures were then subjected to a biological assay to validate their activities. In summary, we sacrificed the diversity of leads in exchange for the efficiency of screening. The DV envelope protein is 495 amino acids in length, forms oligomers, and, along with the M protein, constitutes most of the accessible virion surface that is covered by the envelope membrane. The E protein is responsible for activating “membrane fusion”, the central molecular event during the entry of enveloped RNA viruses into host cells. 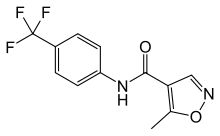 The Dengue virus enters a host cell when the E protein binds to the virus receptor on the host cell surface and activates its conformational rearrangement, causing the E protein in its dimeric pre-fusion form to transform into a trimeric post-fusion structure. This essentially irreversible conformational change induces the fusion between the viral envelope membrane and the host cell membrane, allowing entry to be completed. In short, the DV E protein mediates host cell binding and is essential for infection via a conformationinduced membrane fusion event between the host cell and the virion. In addition, it is also the primary antigen that induces protective immunity and the major antigen for virus neutralization. The crystal structures of the E protein of DV type 2 in both the presence and absence of a bound ligand were deposited in the Protein Data Bank {PDB codes 1oke and 1ok8, respectively; Figure 1). The key difference between these two structures is a local rearrangement of the “kl” b-hairpin and the concomitant opening up of a hydrophobic pocket for ligand binding. For example, the detergent noctyl-b-D-glucoside can occupy this pocket. Mutations that affect the pH threshold for membrane fusion have also been mapped to this hydrophobic pocket. Therefore, Modis et al. proposed that this pocket was a hinge point in the fusionactivating conformational change. This concept made the utilization of structure-based VS to identify inhibitors of DV infection plausible. Therefore, in this study, a well-developed HhAntag691 docking tool, GEMDOCK, was utilized to perform VS on the Comprehensive Medicinal Chemistry databasefor substances thatcoulddock in this hydrophobic pocket of E proteins.
The Dengue virus enters a host cell when the E protein binds to the virus receptor on the host cell surface and activates its conformational rearrangement, causing the E protein in its dimeric pre-fusion form to transform into a trimeric post-fusion structure. This essentially irreversible conformational change induces the fusion between the viral envelope membrane and the host cell membrane, allowing entry to be completed. In short, the DV E protein mediates host cell binding and is essential for infection via a conformationinduced membrane fusion event between the host cell and the virion. In addition, it is also the primary antigen that induces protective immunity and the major antigen for virus neutralization. The crystal structures of the E protein of DV type 2 in both the presence and absence of a bound ligand were deposited in the Protein Data Bank {PDB codes 1oke and 1ok8, respectively; Figure 1). The key difference between these two structures is a local rearrangement of the “kl” b-hairpin and the concomitant opening up of a hydrophobic pocket for ligand binding. For example, the detergent noctyl-b-D-glucoside can occupy this pocket. Mutations that affect the pH threshold for membrane fusion have also been mapped to this hydrophobic pocket. Therefore, Modis et al. proposed that this pocket was a hinge point in the fusionactivating conformational change. This concept made the utilization of structure-based VS to identify inhibitors of DV infection plausible. Therefore, in this study, a well-developed HhAntag691 docking tool, GEMDOCK, was utilized to perform VS on the Comprehensive Medicinal Chemistry databasefor substances thatcoulddock in this hydrophobic pocket of E proteins.
Combination therapies in hematological malignancies is also underscored by their ability to target tumor environment
Tumor microenvironment is a dominant force in inducing resistance to therapy in multiple malignancies. Tumor microenvironment plays a key role in leukemic stem cell maintenance and in modulating signal transduction and resistance in CML and AML. In conclusion, the combination of bortezomib and mitotic inhibitors such as paclitaxel, docetaxel, vincristine or BI 2536 is an effective strategy for targeting of both TKIs -resistant and sensitive Bcr-Abl-positive leukemic cells. These regimens are able to inhibit Bcr-Abl activity and its downstream signaling, and to activate caspase-dependent cell death. In addition, these regimens are able to overcome the resistance to imatinib, dasatinib and nilotinib, brought about by Bcr-Abl protein overexpression or Bcr-Abl mutations, making them attractive potential therapies for Bcr-Abl-positive leukemias such as CML, especially for those resistant to current treatments. As the combined treatment is also efficient in non-CML Bcr-Abl positive cells such as the Baf3 Bcr-Abl cell line, it may also be a promising therapy for non-CML Ph+ leukemias. The initiation of DNA replication is temporally divided into two phases during the cell cycle. First, an inactive form of the replicative MCM helicase is loaded onto origin DNA in G1 phase and then activated upon entry into and during S phase by two sets of kinases: cyclindependent kinase and Dbf4-dependent kinase. DDK is a two-subunit Ser/Thr kinase composed of the Cdc7 kinase and Dbf4 regulatory subunits. DDK mediated phosphorylation of the six-subunit Mcm2-7 helicase is thought to bring about a conformational change in its structure leading to helicase activation. MCM activation is followed by localized DNA unwinding, recruitment of the replisome machinery and the initiation of bi-directional DNA synthesis. Other functions of DDK include facilitation of chromosomal segregation in mitosis and meiosis, the initiation of meiotic recombination, and activation of DNA R428 repair pathways including trans-lesion DNA repair. Cdc7 kinase activity depends on association with its regulatory subunit, Dbf4. Dbf4 is a cell cycle regulated protein whose Ibrutinib Src-bcr-Abl inhibitor abundance peaks during S-phase and then is degraded by end of mitosis. Interaction with Dbf4 is necessary for Cdc7 ATP binding and substrate recognition. Like all protein kinases, the DDK crystal structure reveals an active site in a deep cleft between the N- and C-terminal lobes. The Dbf4 Zn-finger binds to the N-terminal lobe of DDK and is necessary for human DDK activity but is not essential for budding or fission yeast DDK kinase activity. Dbf4 motif M enhances its association with the Cdc7 subunit and is required for the full activity of the kinase in yeast and humans. DDK phosphorylates multiple subunits of the MCM helicase and a recent study in budding yeast indicates that Cdc7 and Dbf4 physically interact with distinct subunits of the Mcm2-7 complex. DDK is over expressed in a number of primary tumors and tumor cell lines. DDK over expression has also been associated with poor prognosis in breast cancers, advanced clinical stage in ovarian carcinoma, and with aggressive phenotype in papillary thyroid carcinomas. Regulating the levels of DDK in tumor cells is an attractive tumor therapeutic strategy. Using neutralizing antibodies, Hunter and colleagues were the first to show that DDK depletion leads to severe disruption of DNA replication in HeLa cells. Using small interfering 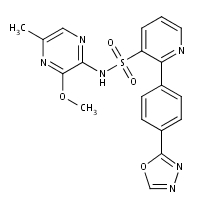 RNAs, Santocanale and colleagues further showed that DDK depletion led to p53-independent apoptosis in HeLa cells whereas a normal human dermal fibroblast cell line underwent a reversible cell-cycle arrest. HeLa cells were unable to arrest at the G1-S phase transition, progressing through a lethal S phase resulting in cell death via apoptosis. This finding has been corroborated in a number of different cell lines. Importantly, tumor cell death induced by depletion of DDK is not accompanied by the induction of known checkpoint markers.
RNAs, Santocanale and colleagues further showed that DDK depletion led to p53-independent apoptosis in HeLa cells whereas a normal human dermal fibroblast cell line underwent a reversible cell-cycle arrest. HeLa cells were unable to arrest at the G1-S phase transition, progressing through a lethal S phase resulting in cell death via apoptosis. This finding has been corroborated in a number of different cell lines. Importantly, tumor cell death induced by depletion of DDK is not accompanied by the induction of known checkpoint markers.
Gliomas are the most common form of primary brain cancer and represent what is currently a generally incurable tumor
In the process of liver fibrosis, stimulation of hepatocyte regeneration and inhibition of apoptosis is essential to treating hepatic fibrosis. In the present study, DAPT treatment was found not to inhibit hepatocyte proliferation. In contrast, DAPT was found likely to inhibit hepatocyte apoptosis to some degree in vivo. We also found that the expression of TGF-b1 was upregulated in the fibrotic livers, and some hepatocytes close to the fibrotic area expressed high levels of TGF-b1, which can induce apoptosis in hepatocytes and stimulate ECM deposition in hepatic fibrosis. We infer that one potential mechanism underlying the protecting effect of DAPT may involve the suppression of TGFb1 expression, which contributes to hepatocyte proliferation and protects hepatocytes from apoptosis. Inhibiting c-secretase can prevent the cleavage of the Notch receptor, blocking Notch signal transduction. The clinical trials with c-secretase inhibitors have revealed several adverse events, such as gastrointestinal toxicity. Other approaches that target the Notch signaling were recently evaluated and showed promising effects accompanied by a lack of intestinal toxicity in preclinical models. These studies shed light on the clinical implications of c-secretase inhibitor. In summary, the present investigation indicates that the Notch signaling pathways become activated in a rat model of liver fibrosis induced by CCl4 and that inhibition of Notch signaling LY2109761 exerts potent anti-fibrotic effects in preclinical models. Our study provides the first evidence for the striking suppressive effects of DAPT on hepatic fibrosis. These findings suggest that inhibition of Notch signaling might be a novel option for hepatic fibrosis therapy. Cells employ multiple mechanisms to repair or tolerate DNA lesions in order to maintain 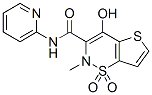 genomic integrity. Translesion DNA synthesis is one of the mechanisms used to tolerate unrepaired DNA lesions. DNA polymerase k is a TLS polymerase that has been shown to catalyze TLS past a variety of DNA lesions, being particularly proficient in the bypass of minor groove N2-dG lesions, including the acrolein-derived adducts c-HOPdG and its ring-opened reduced form, DNA- peptide cross-links, and DNA-DNA interstrand cross-links, as well as adducts induced by activated polycyclic aromatic hydrocarbons such as benzopyrene diolepoxide. Importantly, pol k has been demonstrated to be involved in the tolerance of ICLs induced by a chemotherapeutic agent, mitomycin C. In addition to its role in the bypass of N2-dG lesions, pol k has also been shown to play a role in the processing of various ultraviolet light-induced DNA lesions. Many clinically relevant chemotherapeutic agents, including mitomycin C, cisplatin, and nitrogen mustard, target tumor cells by virtue of their ability to covalently cross-link complementary DNA SJN 2511 446859-33-2 strands, introducing ICLs into the genome. These ICLinducing agents are powerful chemotherapeutic agents as the ICL interferes with vital cellular processes such as DNA replication, RNA transcription, and recombination by preventing transient DNA strand separation. Therefore, although TLS is an essential process for cells to survive genotoxic stress, the ability of pol k to bypass ICLs could limit the efficacy of these agents. Critical to this point are data demonstrating that the effectiveness of mitomycin C was increased when pol k expression was suppressed by siRNA. Germane to these observations, previous reports have suggested that pol k may play a role in glioma development and therefore serve as a potential target for novel routes of therapies.
genomic integrity. Translesion DNA synthesis is one of the mechanisms used to tolerate unrepaired DNA lesions. DNA polymerase k is a TLS polymerase that has been shown to catalyze TLS past a variety of DNA lesions, being particularly proficient in the bypass of minor groove N2-dG lesions, including the acrolein-derived adducts c-HOPdG and its ring-opened reduced form, DNA- peptide cross-links, and DNA-DNA interstrand cross-links, as well as adducts induced by activated polycyclic aromatic hydrocarbons such as benzopyrene diolepoxide. Importantly, pol k has been demonstrated to be involved in the tolerance of ICLs induced by a chemotherapeutic agent, mitomycin C. In addition to its role in the bypass of N2-dG lesions, pol k has also been shown to play a role in the processing of various ultraviolet light-induced DNA lesions. Many clinically relevant chemotherapeutic agents, including mitomycin C, cisplatin, and nitrogen mustard, target tumor cells by virtue of their ability to covalently cross-link complementary DNA SJN 2511 446859-33-2 strands, introducing ICLs into the genome. These ICLinducing agents are powerful chemotherapeutic agents as the ICL interferes with vital cellular processes such as DNA replication, RNA transcription, and recombination by preventing transient DNA strand separation. Therefore, although TLS is an essential process for cells to survive genotoxic stress, the ability of pol k to bypass ICLs could limit the efficacy of these agents. Critical to this point are data demonstrating that the effectiveness of mitomycin C was increased when pol k expression was suppressed by siRNA. Germane to these observations, previous reports have suggested that pol k may play a role in glioma development and therefore serve as a potential target for novel routes of therapies.
Associated with a delta free-energy decrease similar to that observed in the wild-type model
With the polar uncharged Q168, and the replacement of R123 with the polar T123 can thus abrogate these key structural salt bridges, potentially altering the active site conformation of NS3 protease, and in turn impact the HCV-3 sensitivity to PIs. Furthermore, HCV-3, together with HCV-2-4-5 genotypes, also presented two minor RAMs as natural polymorphisms, known to confer low-level resistance to boceprevir and/or telaprevir in vitro. Interestingly, both residues 36 and 175 are located near the protease catalytic domain of HCV NS3, but not close to the boceprevir and telaprevir binding sites in their respective complexes with HCV NS3-NS4 protease. Probably, even if mutations at position 36 and 175 should not be directly involved in resistance to PIs, they can influence the viral replication capacity. For instance, viruses with mutations V36A/ L/M demonstrated a comparable fitness to wild type reference virus. However, since no crystallized structures are to date available for non-1 HCV proteases, the overall impact of such polymorphisms on the three-dimensional protein structure will need further investigations. It is important to mention that very recent data demonstrated a pan-genotypic activity of the second generation macrocyclic PI MK-5172, even against HCV-3 MLN4924 genotype. Furthermore, MK-5172 retained activity also against HCV-1 viral strains harbouring key first generation PI RAMs, thus providing a great opportunity for patients infected with all different HCV-genotypes, including those without virological response to previous regimens. Beside HCV-3, also other genotypes showed remarkable sequence differences from HCV-1b. Of particular interest were those genotype-specific amino acid variations affecting residues associated to macrocyclic and linear PIs-resistance or located in proximity of the PI-binding pocket. For instance, HCV-1a and HCV-1b consensus sequences showed different wild-type amino acids at 17/181 NS3protease positions, including some associated with resistance, Rapamycin cost enhanced replication or compensatory effects if mutated. This amino acidic variability may potentially facilitate viral breakthrough and selection of specific resistant variants, that have been indeed observed consistently more frequently in patients infected with HCV-1a than HCV-1b, using both linear and macrocyclic PIs. On the other hand, according to our GBPM structural analysis, highly conserved  NS3-protease positions among all HCV genotypes were those pivotal for enzyme functionality and stability, such as the catalytic-triad, the oxyanion hole at G137 and the residues involved in Zn2+ binding, and also comprised the majority of residues essential for boceprevir-binding. Interestingly, we also observed two highly conserved stretches encompassing NS3 positions 135-142 and 154-159 that could assist in the rational design of new HCV inhibitors with more favourable resistance profiles. A correlation among conserved NS3 amino acid residues and base-paired organization on the putative RNA secondary structure was also observed. Indeed, highly conserved positions at both amino acid and nucleotide levels were located in highly stable RNA paired stems. Probably, the requirement for base-pairing in these structures severely limits the number of “neutral” sites in the genome, constraining neutral HCV drift, since even synonymous mutations could potentially affect and disrupt the RNA-folding. Interestingly, in our predicted RNA structure model, the conserved codon for resistance-associated residue A156 was base-paired with the conserved codon for residue I153. The presence of RAMs at this position, associated to resistance to all linear and some macrocyclic PIs, did not perturb the overall RNA structural conformation.
NS3-protease positions among all HCV genotypes were those pivotal for enzyme functionality and stability, such as the catalytic-triad, the oxyanion hole at G137 and the residues involved in Zn2+ binding, and also comprised the majority of residues essential for boceprevir-binding. Interestingly, we also observed two highly conserved stretches encompassing NS3 positions 135-142 and 154-159 that could assist in the rational design of new HCV inhibitors with more favourable resistance profiles. A correlation among conserved NS3 amino acid residues and base-paired organization on the putative RNA secondary structure was also observed. Indeed, highly conserved positions at both amino acid and nucleotide levels were located in highly stable RNA paired stems. Probably, the requirement for base-pairing in these structures severely limits the number of “neutral” sites in the genome, constraining neutral HCV drift, since even synonymous mutations could potentially affect and disrupt the RNA-folding. Interestingly, in our predicted RNA structure model, the conserved codon for resistance-associated residue A156 was base-paired with the conserved codon for residue I153. The presence of RAMs at this position, associated to resistance to all linear and some macrocyclic PIs, did not perturb the overall RNA structural conformation.
The catalytic center is suitability of a cancer type for treatment with DNMT and/or HDAC inhibitors in the clinic
Protein C inhibitor is a serine protease inhibitor and a member of the serpin superfamily. PCI has originally been described as a plasma inhibitor of activated protein C. Later, the inhibition of several other proteases, including the pancreatic enzymes trypsin and chymotrypsin, by PCI has been shown.. Like other members of the serpin family, PCI acts as a suicide substrate for its target proteases. Serpins have an exposed reactive center loop which offers a potential cleavage site for the protease. The protease recognizes this sequence and binds to the serpin, forming a reversible Michaelis-like complex. Then the protease cleaves the reactive site peptide bond and the serpin incorporates the RCL into b-sheet A, producing a covalent serpin-protease complex. The enzymeinhibitor complex can dissociate, leaving behind an active protease and a cleaved, inactive serpin. Heparin and other glycosaminoglycans can modify the activity and target enzyme specificity of PCI. The heparin-binding site is a basic patch on helix H, which lies close to the reactive center loop. Heparin changes the 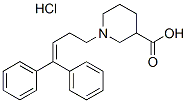 charge of this area, thereby modifying the affinity of PCI towards different proteases. Heparin stimulates the inhibition of APC and thrombin, but abolishes the inhibition of tissue kallikrein by PCI. Antithrombin, GDC-0879 another heparin-binding serpin, uses a different mechanism. Both low molecular weight and unfractionated heparin bind to helix D. This binding leads to a conformational change of AT and an additional part of the reactive center loop is exposed. This results in increased inhibition of coagulation proteases. UFH is furthermore big enough to span from helix D to the protease. It thereby forms a template for AT and thrombin and enhances their interaction. By Northern blotting, a wide tissue distribution of PCI has been demonstrated in humans. PCI mRNA is present in the liver, kidney, heart, brain, lung, spleen, reproductive system and pancreas. Radtke et al. showed by in situ hybridization that PCI is expressed in the exocrine part of the pancreas, and by Western blotting that the protein is present in pancreatic fluid. We have shown that PCI mRNA and protein are also present in keratinocytes of the human skin. Its expression is increased in the more differentiated layers of the epidermis. PCI is also present in several body fluids and secretions, e.g. in plasma and seminal fluid. In rodents, PCI is almost exclusively present in the reproductive tract. This makes it difficult to study the effect of PCI outside the reproductive tract in animal models. Because of its wide tissue distribution, PCI may have several functions in humans. So far, very little is known about these functions. PCI might have a protective effect against cancer progression. Since PCI has affinity for glycosaminoglycans and phospholipids, both Y-27632 dihydrochloride components of the cell membrane, cell membrane association of PCI is not unlikely. We were therefore interested in analyzing the interaction of PCI with serine proteases also present in or on cell membranes. So far there are only a few indications in the literature, suggesting that PCI interacts with type II transmembrane serine proteases. However, as far as inhibition kinetics or the effect of glycosaminoglycans or phospholipids is concerned, no data is available on these interactions. It was therefore the aim of this study to analyze the interaction of PCI with enteropeptidase. EP is a type II transmembrane serine protease, located mainly at the brush border membrane of the epithelial cells of the duodenum and jejunum. Active EP also occurs in duodenal fluid. In the small intestine, EP activates trypsinogen to trypsin. Active human EP is composed of a light and a heavy chain linked by a disulfide bond.
charge of this area, thereby modifying the affinity of PCI towards different proteases. Heparin stimulates the inhibition of APC and thrombin, but abolishes the inhibition of tissue kallikrein by PCI. Antithrombin, GDC-0879 another heparin-binding serpin, uses a different mechanism. Both low molecular weight and unfractionated heparin bind to helix D. This binding leads to a conformational change of AT and an additional part of the reactive center loop is exposed. This results in increased inhibition of coagulation proteases. UFH is furthermore big enough to span from helix D to the protease. It thereby forms a template for AT and thrombin and enhances their interaction. By Northern blotting, a wide tissue distribution of PCI has been demonstrated in humans. PCI mRNA is present in the liver, kidney, heart, brain, lung, spleen, reproductive system and pancreas. Radtke et al. showed by in situ hybridization that PCI is expressed in the exocrine part of the pancreas, and by Western blotting that the protein is present in pancreatic fluid. We have shown that PCI mRNA and protein are also present in keratinocytes of the human skin. Its expression is increased in the more differentiated layers of the epidermis. PCI is also present in several body fluids and secretions, e.g. in plasma and seminal fluid. In rodents, PCI is almost exclusively present in the reproductive tract. This makes it difficult to study the effect of PCI outside the reproductive tract in animal models. Because of its wide tissue distribution, PCI may have several functions in humans. So far, very little is known about these functions. PCI might have a protective effect against cancer progression. Since PCI has affinity for glycosaminoglycans and phospholipids, both Y-27632 dihydrochloride components of the cell membrane, cell membrane association of PCI is not unlikely. We were therefore interested in analyzing the interaction of PCI with serine proteases also present in or on cell membranes. So far there are only a few indications in the literature, suggesting that PCI interacts with type II transmembrane serine proteases. However, as far as inhibition kinetics or the effect of glycosaminoglycans or phospholipids is concerned, no data is available on these interactions. It was therefore the aim of this study to analyze the interaction of PCI with enteropeptidase. EP is a type II transmembrane serine protease, located mainly at the brush border membrane of the epithelial cells of the duodenum and jejunum. Active EP also occurs in duodenal fluid. In the small intestine, EP activates trypsinogen to trypsin. Active human EP is composed of a light and a heavy chain linked by a disulfide bond.